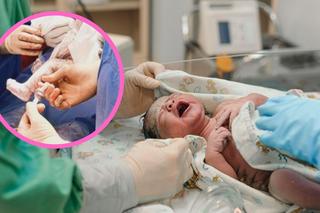









NOWY NUMER
Redaktor prowadząca poleca
TEMAT MIESIĄCA: ŁATWE I WYGODNE KARMIENIE BUTELKĄ

O tym się mówi



Materiał sponsorowany